For U.S. Healthcare Professionals


The Repair study: Effects of macitentan on RV structure and function in pulmonary arterial hypertension1
VIEW THE SERAPHIN CLINICAL DATA to learn more about a large, outcomes-based pivotal trial in PAH.
Important Considerations1
- Data are based on a single-arm, open-label clinical trial and not on a randomized, placebo-controlled clinical trial
- CMR parameters have not been accepted as primary endpoints for pivotal studies in PAH, so these data are hypothesis generating and further research is needed for a better understanding of the significance of CMR parameters as proxy for disease progression
- The open-label design and study size of the Repair study limited subgroup analyses
- OPSUMIT® (macitentan) is an ERA indicated for the treatment of PAH (WHO Group I) (FC II-III) to reduce the risks of disease progression and hospitalization for PAH2
- This study was funded by Actelion Pharmaceuticals Ltd, a part of Johnson & Johnson Innovative Medicine
REPAIR was a 52-week, prospective, multicenter, single-arm, open-label, phase 4 study
Study objectives
- To evaluate the effect of OPSUMIT® on RV and hemodynamic properties in patients with PAH
- Primary endpoints were assessed at Week 26
- To evaluate the safety and tolerability of OPSUMIT® in patients with symptomatic PAH
- Patients’ median exposure time was 52 weeks
Inclusion criteria
- 18 to 74 years of age
- Idiopathic or heritable PAH, PAH related to CTD, drug use or toxin exposure, or simple congenital systemic-to-pulmonary shunts at least 2 years after repair (RHC required to confirm diagnosis)
- Patients who are PAH-treatment naïve or receiving a stable background PDE5 inhibitor for at least 3 months, have a 6MWD of ≥150 m, and a WHO FC I-III
Study population
- Primary interim analysis set: n=42*
- Final analysis set: n=71†‡
- Safety set: n=87§ǁ
Exclusion criteria¶
- Prior use of ERAs, stimulators of soluble guanylate cyclase, or prostacyclin/prostacyclin analogs
The primary endpoints in REPAIR were RVSV (assessed by CMR) and PVR (measured by RHC)1
| Primary endpoints: Change from baseline to Week 26 |
||
|---|---|---|
| RVSV assessed by CMR | ||
| PVR measured by RHC | ||
| Secondary endpoints: Change from baseline to Week 26 |
||
|---|---|---|
| RVESV, RVEDV, RVEF, and RV mass assessed by CMR | ||
| 6MWD | ||
| WHO FC | ||
The single-arm, open-label design and study size of the Repair study limited subgroup analyses.
Patient demographics1
Baseline characteristics (finals analysis set, n=71)
80.3%
(n=57)
45
years
I:
1.4%
(n=1)
II:
47.9%
(n=34)
III:
50.7%
(n=36)
411.2
meters
Etiology


Monotherapy and combination therapy
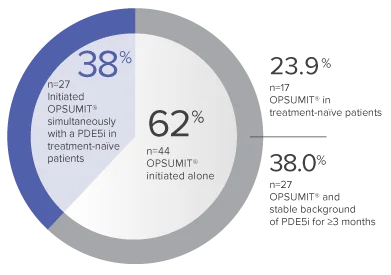
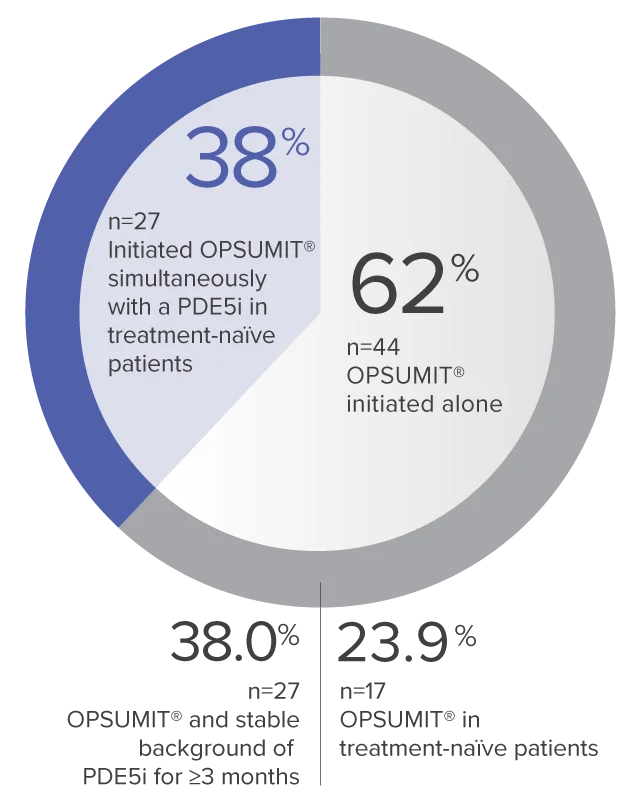
The single-arm, open-label design and study size of the Repair study limited subgroup analyses.
REPAIR Primary Endpoints: RVSV and PVR at Week 26
- The primary interim analysis set (n=42) was declared positive and enrollment was stopped at Week 26 as both primary endpoints (RVSV and PVR) were met
- The final analysis set (n=71) was consistent with the primary interim analysis set
Primary interim analysis set
(n=42)
RVSV
increase
Change from baseline
to Week 26††
LS mean (96% CL)
15.2 (9.3-21.0) mL
Baseline (mean±SD):
50.7±17.5 mL
PVR
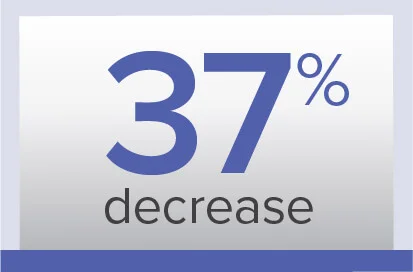
Week 26/baseline ratio‡‡
Geometric mean (99% CL)
0.63 (0.54-0.74) dyn·sec/cm5
Baseline (mean±SD):
900.2±457.6 dyn·sec/cm5
Final analysis set
(n=71)
RVSV
increase
Change from baseline
to Week 26††
LS mean (96% CL)
12.0 (8.4-15.6) mL
Baseline (mean±SD):
52.2±17.2 mL
PVR
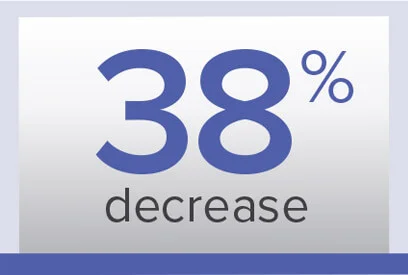
Week 26/baseline ratio‡‡
Geometric mean (99% CL)
0.62 (0.56-0.69) dyn·sec/cm5
Baseline (mean±SD):
974.6±679.0 dyn·sec/cm5
CMR parameters have not been accepted as primary endpoints for pivotal studies in PAH, so these data are hypothesis generating and further research is needed for a better understanding of the significance of CMR parameters as proxy for disease progression.
Observations of RV stroke volume, ejection fraction, and mass at Week 261§§
Final analysis set (n=70)
Change from baseline to Week 26‖‖ in RV parameters
LS mean (95% CL)
(mL)
–16.1
(–20.0 to –12.2)
Baseline (mean±SD):
90.2±40.6
(mL)
–6.2
(–12.8 to 0.4)
Baseline (mean±SD):
149.8±49.1
(%)
+10.6
(7.9 to 13.3)
Baseline (mean±SD):
37.7±14.3
(g)
–10.5
(–14.0 to –7.1)
Baseline (mean±SD):
110.4±47.5
Observations of 6MWD and WHO FC at Week 261
Exercise capacity at Week 26 (n=71)
| Baseline (mean±SD) |
Change from baseline to Week 26## LS mean (95% CL) |
|
|---|---|---|
| 6MWD (m) | 411.2±120.5 | 35.6 (19.0-52.3) |
FC at Week 26 (n=70)
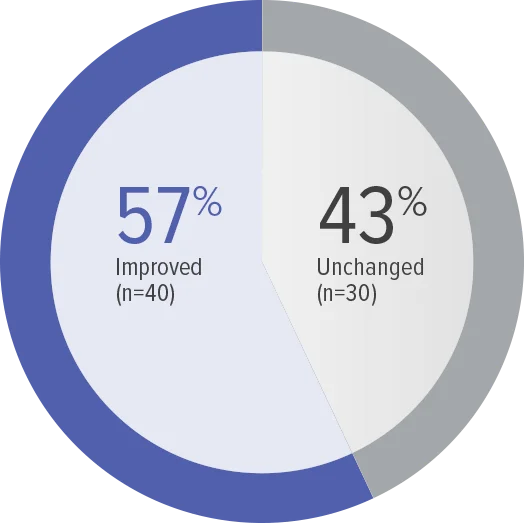
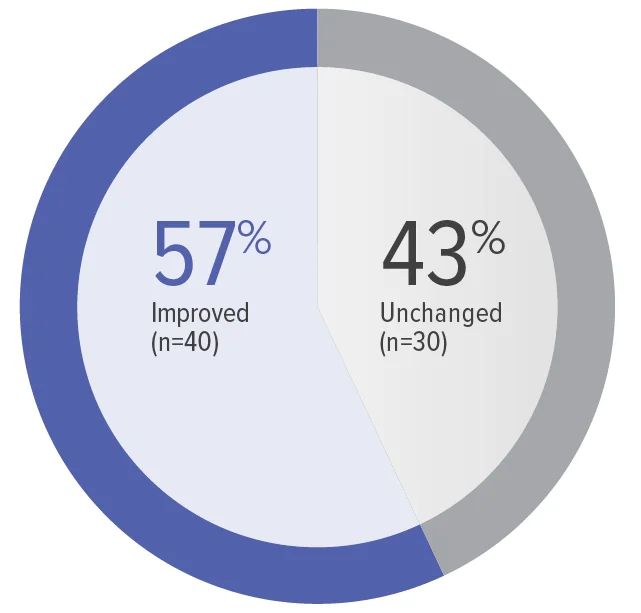
Zero patients worsened in WHO FC.
Baseline WHO FC#: WHO FC I, 1 (1.4%);
WHO FC II, 34 (48.6%); WHO FC III, 35 (50.0%).
Secondary efficacy analyses were performed with no correction for multiple testing; thus, these analyses are of an exploratory nature.
Safety and tolerability in the Repair study
Adverse events observed in the safety set (n=87)
Treatment-emergent adverse events
| Patients with ≥1 treatment-emergent adverse event in ≥10% of patients, n (%) | 75 (86.2) |
|---|---|
| Peripheral edema | 19 (21.8) |
| Headache | 18 (20.7) |
| Dizziness | 12 (13.8) |
| Cough | 10 (11.5) |
| Hemoglobin decreased | 10 (11.5) |
| Upper respiratory tract infection | 10 (11.5) |
| Myalgia | 9 (10.3) |
Adverse events leading to discontinuation of study treatment
| Patients with ≥1 adverse event leading to discontinuation of study treatment, n (%) | 7 (8.0) |
|---|---|
| Aspartate aminotransferase increased | 2 (2.3) |
| Transaminases increased | 2 (2.3) |
| Hypersensitivity | 1 (1.1) |
| Liver function test increased | 1 (1.1) |
| Edema peripheral | 1 (1.1) |
Select treatment-emergent serious adverse events***
| Patients with ≥1 treatment-emergent serious adverse event, n (%) | 14 (16.1) |
|---|---|
| Pneumonia | 3 (3.4) |
| Acute myocardial infarction | 2 (2.3) |
| Pulmonary arterial hypertension | 2 (2.3) |
| Pulmonary embolism | 2 (2.3) |
| Sepsis | 2 (2.3) |
| Cardiac arrest (1 death recorded was the result of a fatal cardiac arrest, which occurred after the patient experienced a pulmonary embolism) | 1 (1.1) |
- Laboratory abnormalities of ALT/AST ≥3 x the ULN were reported for 5 (5.8%) patients in the safety set1
7-Year Data
VIEW RESULTSSafety and Tolerability
LEARN MORE*Primary results were based on the interim analysis set: Prespecified set of the first 42 patients who received at least 1 dose of OPSUMIT® and had valid measurements for both primary endpoints at baseline and at Week 26.
†71 patients with both RVSV and PVR measures at baseline and Week 26.
‡Final analysis set: All enrolled patients who received at least 1 dose of OPSUMIT® and had valid measurement for both primary endpoints at baseline and at Week 26.
§87 patients received ≥1 dose of OPSUMIT®.
ǁSafety set: All screened patients who received at least 1 dose of OPSUMIT®.
¶Reference supplemental methods for a complete list of the exclusion criteria.
#OPSUMIT® is only indicated in WHO FC II and III.
**Only simple congenital systemic-to-pulmonary shunts at ≥2 years postsurgical repair.
††Adjusted change using an ANCOVA model with a factor for PAH-targeted background therapy and a covariate for baseline parameter value.
‡‡Adjusted change using an ANCOVA model with a factor for PAH-targeted background therapy and a covariate for baseline log PVR.
§§Not adjusted for multiplicity.
ǁǁAnalyzed using an ANCOVA with a factor for PAH-targeted background therapy and a covariate for baseline parameter value.
¶¶From pulmonary artery flow.
##From ANCOVA model on parameter change from baseline with factors for PAH-targeted treatment strategy, baseline WHO FC, and parameter at baseline as a covariate.
***Reference supplemental table 6 for a complete list of treatment-emergent serious adverse events.
This site is intended for U.S.
healthcare professionals

You are now leaving OpsumitHCP.com
Clicking CONTINUE below will take you to the selected site, the content for which Johnson & Johnson is not responsible and to which this Privacy Policy does not apply. We encourage you to read the Privacy Policy of every online service you visit.
You are now leaving OpsumitHCP.com
Clicking CONTINUE below will take you to the selected site, the content for which Johnson & Johnson is not responsible and to which this Privacy Policy does not apply. We encourage you to read the Privacy Policy of every online service you visit.
You are now leaving OpsumitHCP.com
Clicking CONTINUE below will take you to the selected site, the content for which Johnson & Johnson is not responsible and to which this Privacy Policy does not apply. We encourage you to read the Privacy Policy of every online service you visit.
You are now leaving OpsumitHCP.com
By clicking on “Continue” below, you will leave OpsumitHCP.com and be directed to the Janssen CarePath site.
You are now leaving OpsumitHCP.com
By clicking on “Continue” below, you will leave OpsumitHCP.com and be directed to the site for iAssist registration.
You are now leaving OpsumitHCP.com
Clicking CONTINUE below will take you to the selected site, the content for which Johnson & Johnson is not responsible and to which this Privacy Policy does not apply. We encourage you to read the Privacy Policy of every online service you visit.
You are now leaving OpsumitHCP.com
Clicking CONTINUE below will take you to the selected site, the content for which Johnson & Johnson is not responsible and to which this Privacy Policy does not apply. We encourage you to read the Privacy Policy of every online service you visit.
You are now leaving OpsumitHCP.com
Clicking CONTINUE below will take you to the selected site, the content for which Johnson & Johnson is not responsible and to which this Privacy Policy does not apply. We encourage you to read the Privacy Policy of every online service you visit.
You are now leaving OpsumitHCP.com
Clicking CONTINUE below will take you to the selected site, the content for which Johnson & Johnson is not responsible and to which this Privacy Policy does not apply. We encourage you to read the Privacy Policy of every online service you visit.
You are now leaving OpsumitHCP.com
Clicking CONTINUE below will take you to the selected site, the content for which Johnson & Johnson is not responsible and to which this Privacy Policy does not apply. We encourage you to read the Privacy Policy of every online service you visit.